
Inteligencia artificial explicable para el tratamiento de la leucemia mieloide aguda: Un enfoque híbrido
Quiero compartirles un análisis que realicé, enfocado en el uso de inteligencia artificial explicable para apoyar la toma de decisiones médicas en casos de leucemia mieloide aguda (LMA).
El objetivo principal es combinar interpretabilidad y buen rendimiento predictivo, utilizando modelos de Explainable Boosting Machine (EBM) y XGBoost, complementados con explicaciones de SHAP.
El desafío de la leucemia mieloide aguda (LMA)
La leucemia mieloide aguda (LMA) es un tipo de cáncer hematológico que se origina en la médula ósea. En esta enfermedad, los mieloblastos, células precursoras de la línea mieloide, dejan de madurar y se multiplican de forma descontrolada, lo que impide la producción normal de glóbulos rojos, glóbulos blancos y plaquetas. Esto provoca síntomas como anemia, infecciones recurrentes y hemorragias. La LMA progresa rápidamente y representa aproximadamente el 80 % de las leucemias agudas en adultos, siendo más común después de los 60 años.
La alta heterogeneidad clínica y genética de esta enfermedad dificulta la estratificación del riesgo y la elección del tratamiento más adecuado. El diagnóstico suele involucrar exámenes de sangre, biopsia de médula ósea y estudios genéticos. El tratamiento estándar se guía por la clasificación de riesgo de la European LeukemiaNet (ELN), que agrupa a los pacientes en riesgo bajo, intermedio y alto. Sin embargo, los pacientes clasificados como de riesgo intermedio, que representan entre el 40 % y el 50 %, suelen generar incertidumbre terapéutica. En muchos casos, se requieren estudios adicionales, lo que puede retrasar el inicio del tratamiento.
En este contexto, las técnicas de aprendizaje automático han surgido como herramientas prometedoras. Permiten procesar grandes volúmenes de datos clínicos y genómicos para mejorar las predicciones y personalizar el tratamiento. No obstante, muchos modelos son considerados “cajas negras” debido a su falta de transparencia, lo que limita su adopción en entornos médicos. De ahí la necesidad de desarrollar modelos explicables y precisos.
Un enfoque híbrido explicable
Para abordar esta necesidad, se diseñó un sistema híbrido de apoyo a la decisión clínica que integra dos modelos complementarios:
Explainable Boosting Machine (EBM), valorado por su interpretabilidad, que permite analizar la influencia específica de cada variable en la predicción.
Extreme Gradient Boosting (XGBoost), reconocido por su sólido desempeño en tareas de clasificación médica.
La combinación de ambos modelos busca orientar mejor las decisiones terapéuticas, especialmente en los pacientes de riesgo intermedio. Este enfoque se fortaleció mediante un esquema de ensemble con votación ponderada, en el cual se asigna mayor peso al modelo con mejor rendimiento validado. Así se logra mayor robustez y se reduce el riesgo de sobreajuste.
Metodología y fuentes de datos
La implementación se realizó en Python, utilizando librerías como XGBoost, interpret y SHAP. Estas herramientas permitieron construir modelos explicables y visualizar el impacto específico de cada variable sobre la predicción de supervivencia.
Se trabajó con tres fuentes principales de datos:
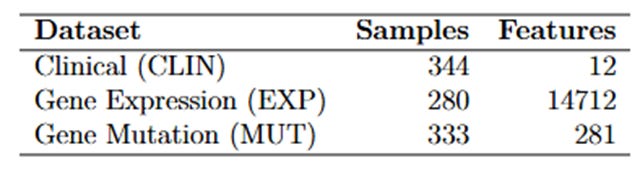

CLIN: Contiene información sobre los niveles de expresión de diversos genes, con valores numéricos que indican la intensidad de su expresión. Los valores positivos y negativos reflejan el aumento o la disminución de la actividad génica, lo que permite realizar análisis para identificar patrones asociados a la leucemia mieloide aguda.
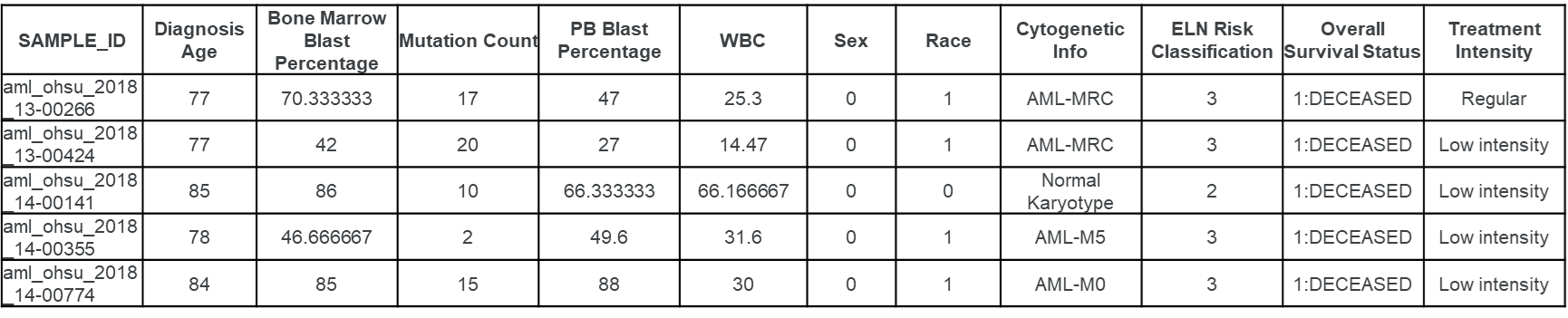
EXP: Contiene información sobre los niveles de expresión de diversos genes, con valores numéricos que indican la intensidad de su expresión. Los valores positivos y negativos reflejan el aumento o la disminución de la actividad génica, lo que permite realizar análisis para identificar patrones asociados a la leucemia mieloide aguda.
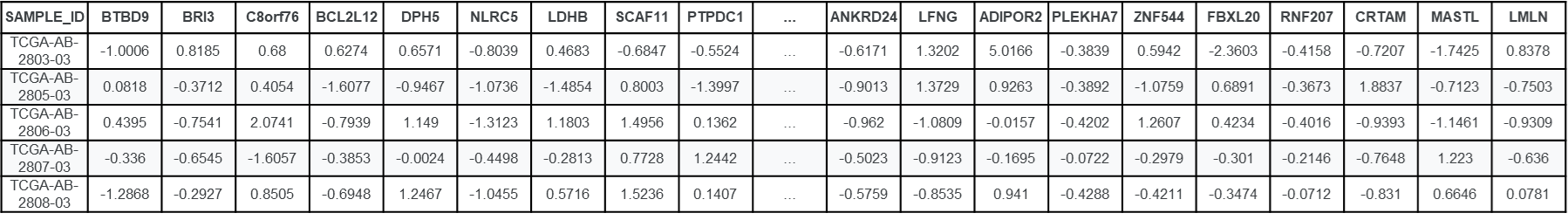
MUT: Reúne información de 333 pacientes, indicando la presencia o ausencia de 281 mutaciones en genes clínicamente relevantes. Entre ellos se destacan genes como FLT3, NPM1, DNMT3A, IDH1, IDH2, TP53, TET2, ASXL1, RUNX1, CEBPA, NRAS, KRAS, SF3B1, U2AF1 y SRSF2, los cuales, según la literatura especializada, desempeñan un papel determinante en el pronóstico y en las decisiones terapéuticas en casos de LMA.
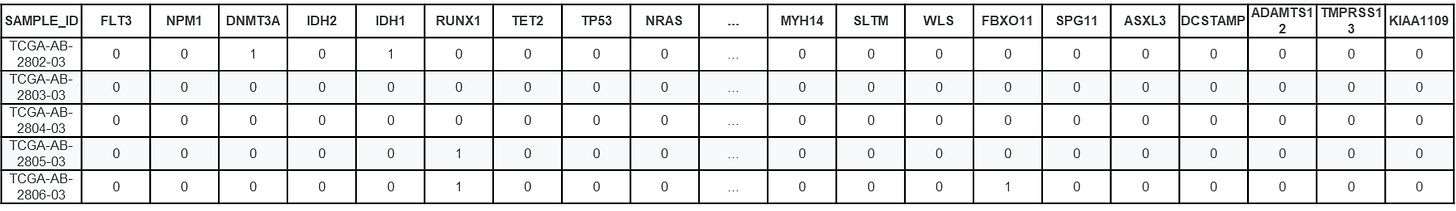
Se inició con el preprocesamiento de los datos para garantizar su integridad y homogeneidad. Se excluyeron pacientes menores de 18 años y aquellos con menos del 20 % de blastos en la médula ósea, ya que este valor forma parte del criterio diagnóstico para leucemia mieloide aguda (LMA). También se eliminaron registros duplicados basados en el campo SAMPLE_ID.
La variable categórica “Overall Survival Status” fue convertida a valores numéricos (0 y 1) mediante LabelEncoder, permitiendo su uso en modelos de clasificación binaria.
Para las variables clínicas continuas, se aplicó un imputador de vecinos más cercanos (KNNImputer) con k = 3, que permite completar valores faltantes utilizando muestras similares. Esto evita la pérdida de pacientes valiosos debido a datos incompletos.
Las variables categóricas como Treatment Intensity, Sex, Race, entre otras, fueron transformadas en variables binarias utilizando codificación one-hot (pd.get_dummies()), una técnica esencial para que los modelos numéricos puedan procesarlas adecuadamente.
Posteriormente, las variables continuas fueron estandarizadas con StandardScaler, normalizando sus valores para que todas presenten media cero y desviación estándar uno. Este procedimiento evita que variables con diferentes escalas influyan de forma desproporcionada en el aprendizaje del modelo.
Se implementaron funciones personalizadas para combinar los conjuntos de datos clínicos con los de mutaciones o expresión génica, utilizando SAMPLE_ID como clave de unión. En el caso de los datos genéticos, también se agregó previamente la variable Treatment Intensity antes de la fusión, permitiendo su análisis conjunto con la información molecular.
Dado el elevado número de variables genéticas, se realizó un proceso de selección de características con el objetivo de conservar únicamente aquellas más relevantes para la predicción del estado de supervivencia.
Selección de mutaciones genéticas (chi-cuadrado): Se aplicó la prueba estadística de chi-cuadrado para evaluar la asociación entre la mutación de cada gen y la variable objetivo. Solo se conservaron las variables con un valor de p < 0.1, lo que indica una relación estadísticamente significativa con la variable a predecir. Este procedimiento permitió reducir la dimensionalidad del conjunto de datos y enfocar el modelo en genes potencialmente relevantes.
Selección de expresión génica (LinearSVC con penalización L1): Para los datos de expresión génica se utilizó un modelo LinearSVC con regularización L1. Este modelo asigna pesos a cada variable (gen), y se seleccionan como relevantes aquellas cuyo coeficiente es distinto de cero. Esta técnica automatiza el proceso de filtrado, eliminando genes con baja o nula capacidad predictiva.
Ambos procesos de selección se realizaron exclusivamente sobre el conjunto de entrenamiento, con el fin de evitar data leakage, es decir, impedir que el modelo tenga acceso previo a datos que posteriormente serán utilizados en su validación.
La evaluación final se llevó a cabo sobre un conjunto de prueba independiente, utilizando métricas como AUC, F1 Score, precisión, recall y exactitud, lo que permitió cuantificar el desempeño de cada modelo y de la estrategia de ensemble.
Se construyeron siete combinaciones posibles de los conjuntos de datos (por ejemplo, CLIN_MUT_EXP, CLIN_EXP, MUT_EXP, etc.) con el fin de evaluar su impacto individual y conjunto en el rendimiento predictivo.
Entrenamiento
Para evaluar el impacto de cada tipo de información en la predicción del estado de supervivencia, se consideraron siete subconjuntos de datos:
CLIN: solo datos clínicos
MUT: datos de mutaciones
EXP: perfiles de expresión génica
CLIN_MUT: datos clínicos y de mutaciones
CLIN_EXP: datos clínicos y de expresión génica
MUT_EXP: datos de mutaciones y expresión génica
CLIN_MUT_EXP: integración de datos clínicos, de mutaciones y de expresión génica
Con el fin de evitar overfitting y garantizar una evaluación más confiable, se utilizó un esquema de división estratificada en cuatro subconjuntos de datos, disponibles en:
https://raw.githubusercontent.com/jdmanzur/ml4aml_databases/main/train_test_split/
Este enfoque permitió comparar el rendimiento de los modelos en distintos niveles de integración de la información, evaluando el valor añadido de los datos clínicos, moleculares y genómicos en la predicción de la supervivencia.
Se utilizaron los siguientes archivos:
X_train_id.csv
X_valid_id.csv
X_test_id.csv
X_final_test_id.csv
Cada archivo contiene los SAMPLE_ID previamente seleccionados, asegurando que la proporción entre las clases sea similar en todos los subconjuntos. La finalidad de cada conjunto es la siguiente:
Entrenamiento: se utilizó para entrenar los modelos.
Validación: sirvió para ajustar los hiperparámetros y seleccionar el mejor modelo.
Prueba: se destinó a la evaluación final del rendimiento.
Prueba final: se empleó para generar resultados replicables en artículos científicos o presentaciones.
EBM: Se utilizó la librería interpret.glassbox para entrenar el modelo con los datos procesados del conjunto de entrenamiento. No fue necesario aplicar ningún tipo de codificación especial, ya que este modelo puede trabajar directamente con variables numéricas y categóricas ya codificadas. Se aplicó validación cruzada k-fold con k = 5, exclusivamente sobre el conjunto de entrenamiento.
XGBoost: El modelo fue entrenado inicialmente con los parámetros por defecto. Posteriormente, se realizó una búsqueda de hiperparámetros utilizando validación cruzada estratificada mediante StratifiedKFold (k = 5), combinada con GridSearchCV. Esta estrategia permitió explorar diferentes configuraciones de manera robusta sin utilizar el conjunto de validación, preservando así su integridad como conjunto de evaluación ciega. La mejor combinación de hiperparámetros identificada se utilizó para entrenar el modelo final, que fue evaluado en los conjuntos de validación y prueba.
Modelo de ensemble: Se construyó a partir de la combinación de las predicciones de EBM y XGBoost, asignando un mayor peso al modelo que obtuvo mejores resultados en términos de F1 Score durante la validación.
La fórmula del modelo de ensemble se expresa como:
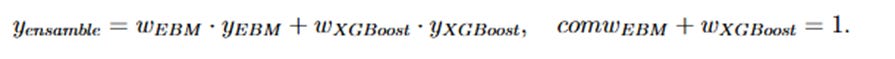
Resultados
Los modelos ensemble superaron en general a los modelos individuales. El subconjunto que arrojó los mejores resultados fue el MUT_EXP, es decir, la combinación de mutaciones genéticas y expresión génica.
En este escenario, el ensemble MUT_EXP alcanzó:
AUC: 0.9167
Precisión: 82.65 %
F1 Score: 0.7902
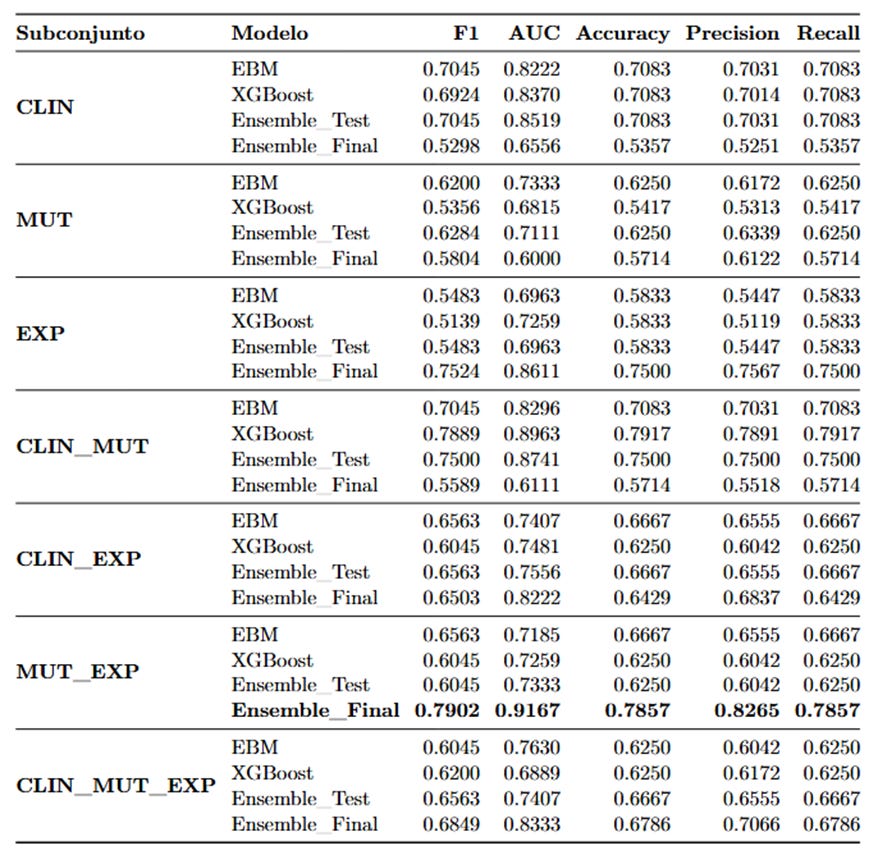
Estos resultados destacan la importancia de integrar información genómica y molecular para lograr predicciones más precisas en pacientes con LMA.
Factores determinantes de la supervivencia
El análisis de interpretabilidad con EBM y los valores SHAP permitieron identificar las variables más influyentes en la predicción de la supervivencia:
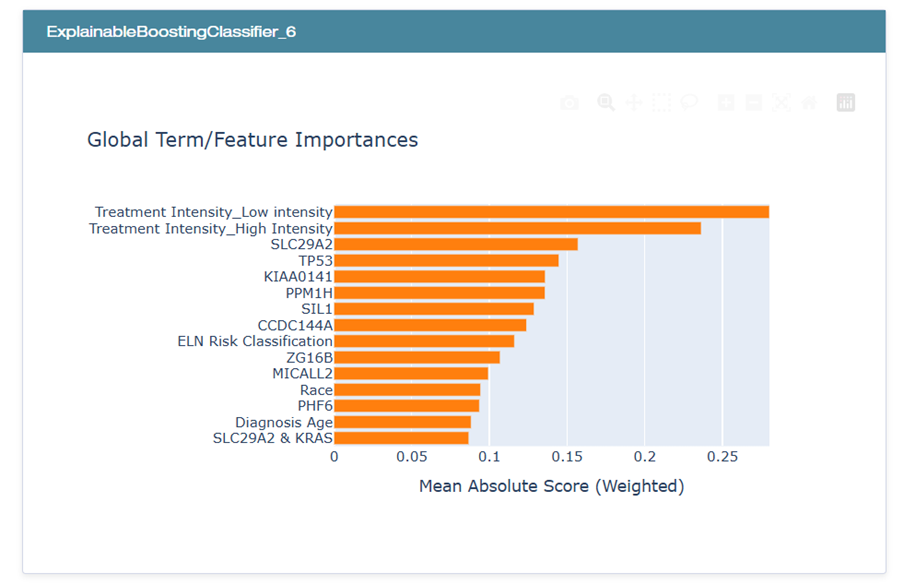
En el gráfico, se evidencia que las variables relacionadas con la intensidad del tratamiento, en particular, Treatment Intensity_Low intensity y Treatment Intensity_High intensity, ocupan las primeras posiciones en términos de relevancia. Les siguen marcadores genéticos como TP53, SLC29A2 y KIAA0141, mientras que atributos clínicos adicionales como Race, Diagnosis Age y ELN Risk Classification contribuyen de forma moderada al desempeño del modelo.
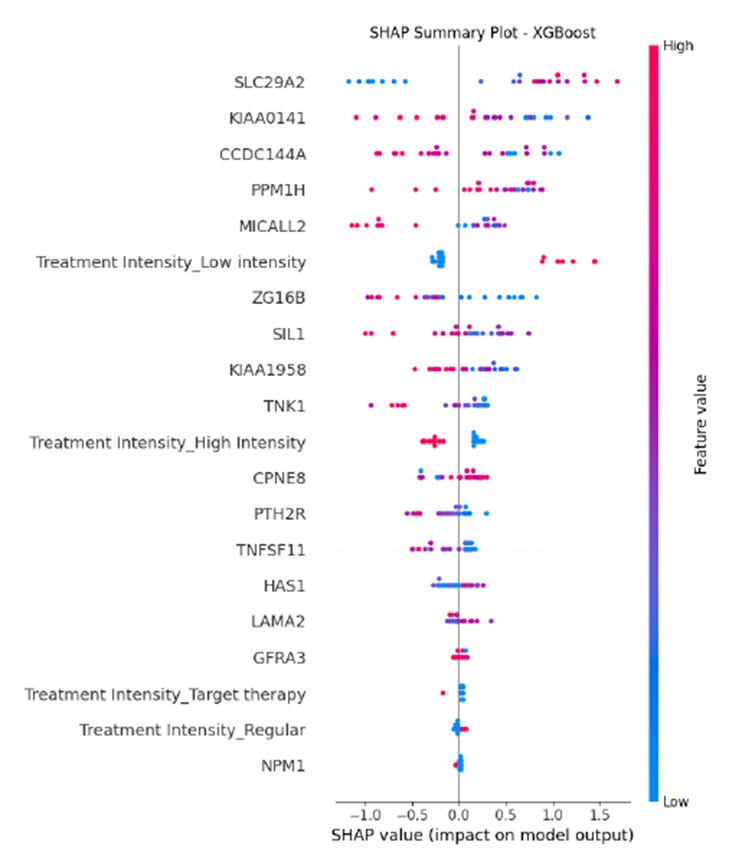
En el diagrama SHAP para XGBoost, cada punto corresponde a una muestra y su color representa el valor de la característica (de bajo, en azul, a alto, en rosa).
El eje horizontal indica el impacto sobre la probabilidad de la clase positiva (valores positivos aumentan dicha probabilidad, mientras que valores negativos la reducen).
Se observa que variables como SLC29A2, KIAA0141 y la intensidad del tratamiento (Treatment Intensity_Low intensity, Treatment Intensity_High intensity y Treatment Intensity_Regular) se destacan entre las de mayor influencia, reflejando la importancia combinada de factores genéticos y clínicos en la predicción del estado de supervivencia.
Conclusiones
Se propuso y evaluó un sistema de apoyo a la decisión terapéutica para pacientes con leucemia mieloide aguda (LMA), basado en un ensemble híbrido explicable que combina los modelos EBM y XGBoost.
El modelo que integró datos de mutaciones genéticas y expresión génica (MUT_EXP) obtuvo un rendimiento destacado en el conjunto de prueba final, con un AUC de 0.9167, una precisión del 82.65 % y un F1 Score de 0.7902.
La solución alcanzó un equilibrio sólido entre precisión e interpretabilidad, generando predicciones confiables y explicables, lo cual es clave para su adopción clínica. Sin embargo, se identificaron limitaciones relacionadas con el tamaño de los datos y la necesidad de validar el modelo en poblaciones más amplias.
El análisis destaca el potencial de los enfoques explicables en inteligencia artificial aplicada a la medicina, sentando las bases para el desarrollo de sistemas de apoyo clínico más confiables y efectivos en el tratamiento de la LMA.